


Medical device companies hoping to bring their products to market in the European Union (EU) will now have to comply with the Medical Device Regulation (MDR), an update from the old Medical Device Directive (MDD). Companies that were already compliant with the MDD need to pay attention to this updated regulation, which departs from the MDD in several key ways.
Let’s dive into more details and compare the key MDD vs. MDR differences — and how Kapstone can partner with you to ensure compliance.
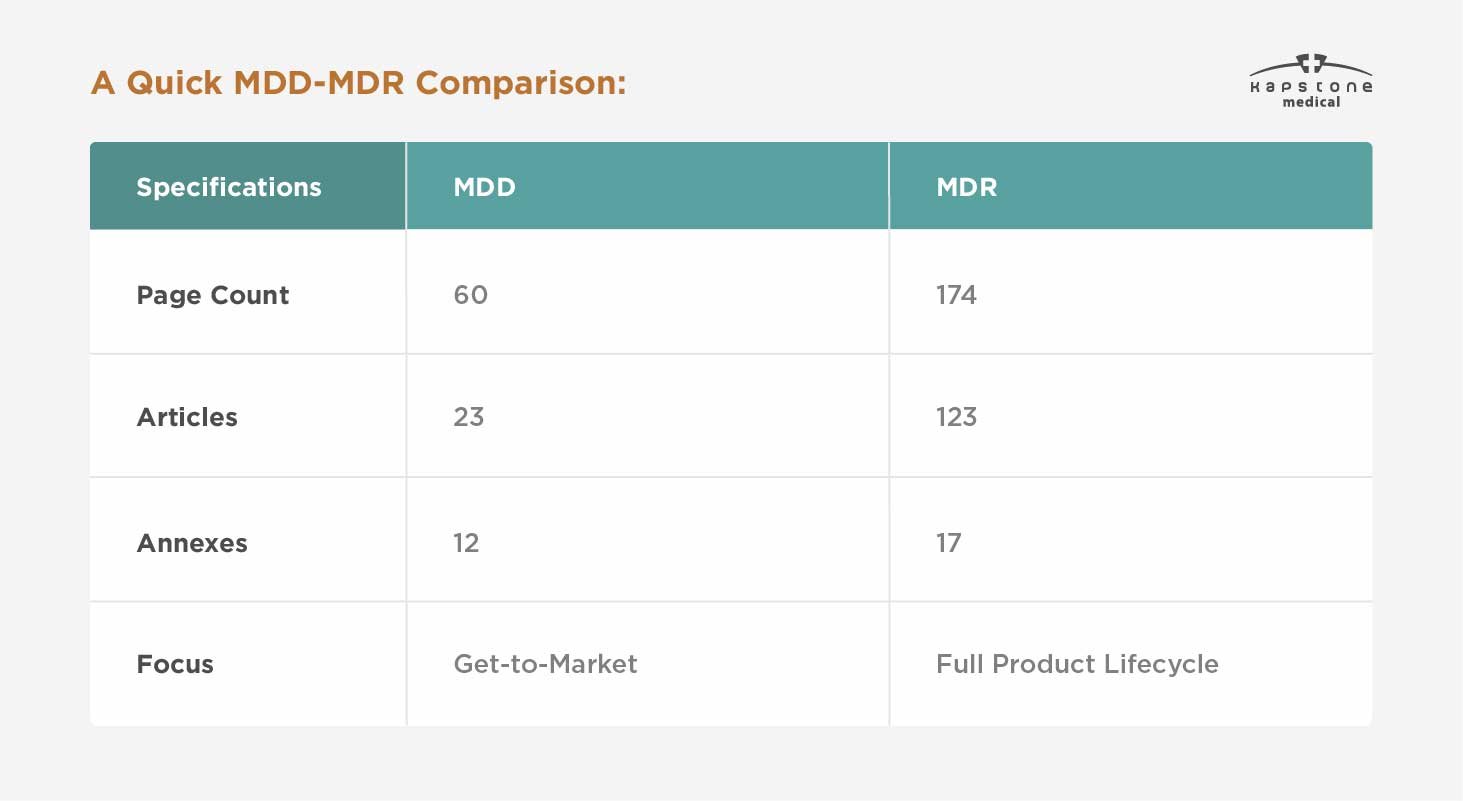
Why was the MDD Updated?
The MDD has been in effect within the EU since 1993. With the drastic changes in the landscape, the MDD is now outdated, leading to the need for a more comprehensive and technology-aware set of regulations.
Software-enabled devices, such as fitness tracking apps, were a long way off when the MDD originated. The MDR accounts for the full technological landscape, establishing guardrails for the regulation, manufacturing, and commercialization of cutting-edge medical devices.
Furthermore, the demographics are trending older across the globe. Thanks to innovation in healthcare, people are living longer — and relying on medicines and medical devices more than ever before.
While the MDD focused on getting a product to market, the MDR expands to consider the full product lifecycle: development, testing, manufacturing, commercialization, efficacy, safety, and long-term use. This is not to say that the MDD ignored those elements of medical device regulation and use. The MDR merely codifies their application in greater detail.
.
Key Difference 1: Unique Device Identification System (UDI)
Each device will need to have a unique device identifier, or UDI. This provides increased tracking abilities throughout the entire supply chain worldwide. All devices except those that are custom made will require this number for better identification and traceability — and, therefore, accountability.
The UDI consists of numbers and letters, and will abide by a global coding standard. The manufacturer is responsible for identifying each device and placing it onto the device itself, so this might be another update that requires an adjustment in standard operating procedures.
Key Difference 2: New Safety Requirements for Medical Devices
Safety evaluations have always been an integral part of medical device development, testing, and initial use. Fundamentally, the MDR expands on the MDD by increasing scrutiny on device safety throughout the full product lifecycle.
Pre-Market Requirements
Medical device development companies hoping to bring their products to market in the EU now must submit additional safety and performance data. While the MDD had 13 Essential Requirements (ERs), the MDR now has 23 General Safety and Performance Requirements (GSPRs). New requirements include:
- Requirements for devices that administer or contain drugs
- Requirements for devices that contain tissues of human or animal origin
- Requirements for disposal
- Requirements for devices to be used by people who are not medical professionals
- General requirements for labeling
Post-Market Surveillance (PMS) Requirements
The MDR holds device manufacturers responsible for all PMS activities. Under this new regulation, manufacturers must:
- Plan, establish, document, implement, maintain, and update a PMS system that is proportionate to a device’s degree of risk and appropriate for its type
- Actively and systemically gather, record, and analyze data on the quality, performance, and safety of a device throughout its lifetime
The purpose of this reporting is to demonstrate that a product’s actual market use mirrors its intended use, risks and risk management have not changed, and device benefits continue to outweigh risks. When changes, risks, complaints, or adverse events are identified, manufacturers are responsible for making the appropriate changes.
Class I devices require only a consistent PMS report, but devices in higher risk classes also require a Periodic Safety Update Report (PSUR). A PSUR should include the results and conclusions of PMS activities and analysis, findings from any post-market clinical follow-up, and sales data. Manufacturers of Class III and implantable devices must also submit their PSUR to the notified body annually.
Finally, the MDR requires that certain serious incidents be reported within 15 days. Under the MDD, the window was 30 days.
Key Difference 3: A Broader Definition of Medical Devices
Under MDR guidance, products that were never previously categorized as “medical devices” now fall under that umbrella, and are subject to all class- and type-appropriate MDR requirements. These products include:
- Contact lenses and other products used in/on the eye
- Products introduced into the body via surgically invasive means to modify the autonomy
- Products and substances used for facial or other subcutaneous fillings
- Equipment used for liposuction, lipolysis, or lipoplasty
- High-intensity radiation equipment used for tattoo and hair removal
- Equipment that stimulates the brain via electrical or magnetic current
- Products intended for cleaning, disinfecting, and sterilizing medical devices
- Products for the control and support of conception
Key Difference 4: New Medical Device Classifications
All medical devices that carry a CE mark will be required to meet the new EU MDR guidelines, and no devices will be grandfathered in.
Given the logistical and financial hurdles that manufacturers will have to overcome to achieve compliance, devices that already carry a valid MDD certificate have a bit of a grace period. However, all MDD certificates become invalid on May 27, 2024.
The MDR also reclassifies several devices into higher risk categories, meaning devices may be subject to increased scrutiny and both pre- and post-market regulation. Example reclassifications include:
- Under MDD, most medical software was classified as Class I. Under MDR, some may fall into higher classifications
- All spinal disk replacement devices, devices that contact the spine, and extremity joint replacement devices will be reclassified from Class IIb to Class III (excluding components such as screws, wedges, plates, and instruments)
- Implantable devices that are Class IIb will be broken into Well Established Technology (WET) and non-WET categories
- Invasive surgical instruments that are reusable have a new classification: Class Ir
Collaboration & Consultation
To navigate the complexities of the MDR, we encourage medical device companies to collaborate with regulatory experts and seek guidance from authorities well versed in such policy language.
That’s where we enter the picture. We’ll work with you throughout the development process to ensure the design, engineering, manufacturing, regulatory, and other crucial considerations are handled.
Would you like to learn more about how we can guide you throughout the process to ensure your devices are hitting every mark? Click the link below to get started.


