


Key Takeaways - Terminology Matters:
The FDA does not "approve" all devices. Using the wrong term (e.g., claiming a Class II device is "approved" rather than "cleared") can be deemed misbranding and get your startup in legal trouble.
- Risk Dictates Pathway: Your path to market is determined by the risk level and classification of your device (Class I, II, or III).
- Registered vs. Cleared vs. Approved:
- Registered: All medical device companies must register their facility and list their devices.
- Cleared (510(k) Premarket Notification): For medium-risk devices (some Class I, and Class II) that are substantially equivalent to an existing FDA-Cleared device.
- Granted (De Novo): for novel low to moderate risk devices (Cass I or Class II) for which no substantially equivalent device exists. This process is known as a request for classification.
- Approved (PMA – Premarket Approval): For high-risk devices (Class III). Generally requires rigorous clinical trials.
- Integrated Approach: Regulatory strategy should not be an afterthought; it must be integrated at inception of your product development process. Doing so will save time and capital.
Introduction: Why Regulatory Terminology Impacts Your Launch
If you are a founder with a medical device concept, you are likely focused on engineering, prototyping, and securing funding. However, your Regulatory Strategy is the invisible hand that controls your timeline and budget.
One of the most common pitfalls we see at Kapstone Medical is confusion around FDA terminology. Investors and the FDA alike expect precision. Knowing which regulatory pathway (e.g. listing only, 510(k), De Novo, or PMA) is appropriate for your device will inform your pitch and your go-to-market plan.
Here is the definitive guide to understanding FDA classifications and what they mean for your medical device startup.
The Foundation: Understanding FDA Device Classes
Before discussing "approval" or "clearance," you must identify where your device fits on the risk spectrum. The FDA classifies devices into three categories based on the risk associated with their intended use/indications for use.
|
Class |
Risk Level |
Examples |
Regulatory Burden |
|
Class I |
Low Risk |
Tongue depressors, bandages, surgical gloves |
Lowest: General Controls, generally 510(k) exempt (with exceptions). Novel Class I devices may require De Novo Grant. |
|
Class II |
Moderate Risk |
Wheelchairs, CT scanners, patient monitors, some surgical instruments |
Medium: Generally requires Premarket Notification 510(k)Clearance. Novel Class II devices may require De Novo Grant. |
|
Class III |
High Risk |
Pacemakers, heart valves, life-sustaining implants |
Highest: Requires Pre-Market Approval (PMA). |
 1. What Does "FDA Registered" vs “Listing” Mean?
1. What Does "FDA Registered" vs “Listing” Mean?
- “Registered” Applies to: The establishment(s) and their owners involved in the production and distribution of a medical device intended to be commercially distributed in the United States. This includes the legally accountable ‘owner’ of the device (manufacturer), and any additional establishments to whom activities are outsourced such as the specification developer, contract manufacturer, distributor, importer, relabeling/repackager, reprocessor, etc. Establishments must register annually.
- “Listing” Applies to a medical device must be listed under a registered facility to indicate that the facility is involved in the production and/or distribution of that device.
- The Gist: It is an administrative requirement, not an endorsement of the device's quality or its compliance. Registering a facility and its associated devices identifies the establishment(s) responsible for the various production activities and thus which establishments require periodic FDA inspection.
Kapstone Insight: While you must register your facility, you cannot market your device as "FDA Registered". Devices get “listed”. Establishments get “Registered”. It is important not to imply that FDA has tested a device. Using the term “FDA Registered Device” is incorrect and can be considered to be adulterated or misbranded product which is a compliance red flag.
2. What Does "FDA Cleared" Mean?
- Applies to: Devices (class I (rare) or class II (most common)) that have been reviewed by FDA, and cleared to commercially distribute product in the US.
- The Mechanism: Pre-market Notification in accordance with Section 510(k) of the Federal Food, Drug, and Cosmetic Act (FDCA)
FDA Clearance is the process for devices that can prove they are "substantially equivalent" to a legally marketed device (known as a "predicate").
When the FDA clears a device, they are saying, "We agree that this new device is at least as safe and effective as the predicate device." A clearance does NOT ensure the device or the establishment(s) are compliant with the Quality Management System Regulations.
How Kapstone Helps: We help founders identify the best "predicate" device early in the development process. By designing your product to align with what the predicate verification and validation activities, we can help streamline your 510(k) submission and accelerate your path to revenue.

3. What Does “Granted” Mean?
- Applies to: Class I or Class II low-to-moderate risk devices that are novel/unique.
- The Mechanism: De Novo request for classification
FDA Grant/Granted indicates that no other low to moderate risk device like yours has been reviewed by FDA and previously marketed. This pathway is used by a manufacturer to request that FDA classify their device when no suitable “predicate” exists. A classification is “Granted”.
To get a device classification “Granted” you must identify the hazards and risks associated with your device’s intended use and demonstrate (via verification and validation testing) the device’s safety and efficacy. This may require clinical studies and/or human factors studies. It is recommended to request a Pre-Submission with FDA to obtain clarity on what FDA expects to be submitted as evidence of safety and efficacy that is unique to your device.
4. What Does "FDA Approved" Mean?
- Applies to: Class III Devices.
- The Mechanism: Pre-Market Approval (PMA).
FDA Approval is the "gold standard" of regulatory review, reserved for high-risk or life-sustaining devices.
To get a device Approved, you must prove—from scratch—that the device is safe and effective. This almost always requires extensive laboratory studies, human clinical trials, and human factors studies. The FDA must give "Approval" before you can market the device.
Summary: Which US FDA Regulatory Path is Right for Your Startup?
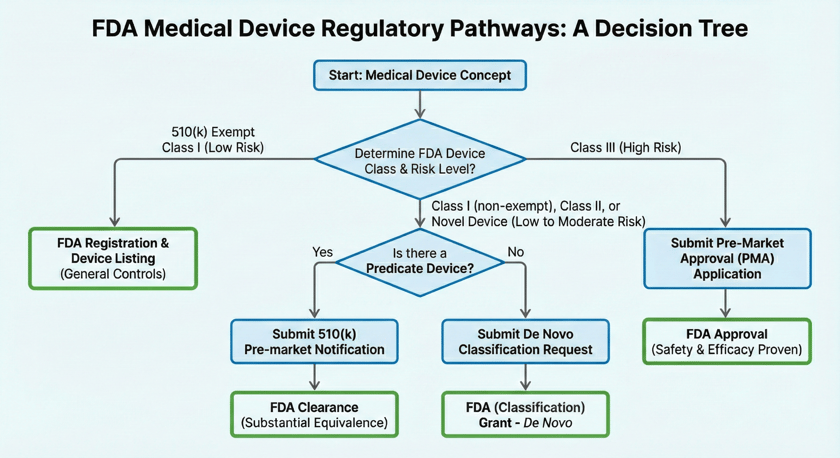
- If you are making a generic surgical tool: You likely just need to apply general controls, Register and List.
- If you are iterating on existing tech (e.g., a new smart catheter) of a low to moderate risk device: You likely need FDA Clearance (510k).
- If you are inventing a novel low to moderate risk device: You likely need a classification grant (De Novo).
- If you are inventing a life-support system: You likely need FDA Approval (PMA).
Partner with Experts to Navigate the Process
At Kapstone Medical, we don’t just handle the paperwork; we integrate regulatory strategy into the design and development of your device. By solving regulatory challenges during the concept stage, we save our clients time and money.
Do you have a product concept and need to determine your regulatory pathway?
Contact Kapstone Medical today for a consultation.

